Methyl-Seq data are derived from Deoxyribonucleic Acid (DNA) molecules that have been isolated from a biological sample (e.g. individual cells or nuclei, cell culture, tissue, organ, or whole organism) then manipulated to distinguish between methylated and unmethylated nucleotides (primarily cytosines), and prepared as libraries to be sequenced on a next generation sequencing platform. Methylated nucleotides are those that have had a methyl group transferred to them through a process known as methylation. This type of epigenetic modification is most widely studied in the context of regulating gene expression.
Some common uses of Methyl-Seq data include:
- identifying individual bases, or regions of bases, that have been methylated in biological samples
- determining how the methylation state of biological samples change in response to a treatment and/or across environmental gradients or time
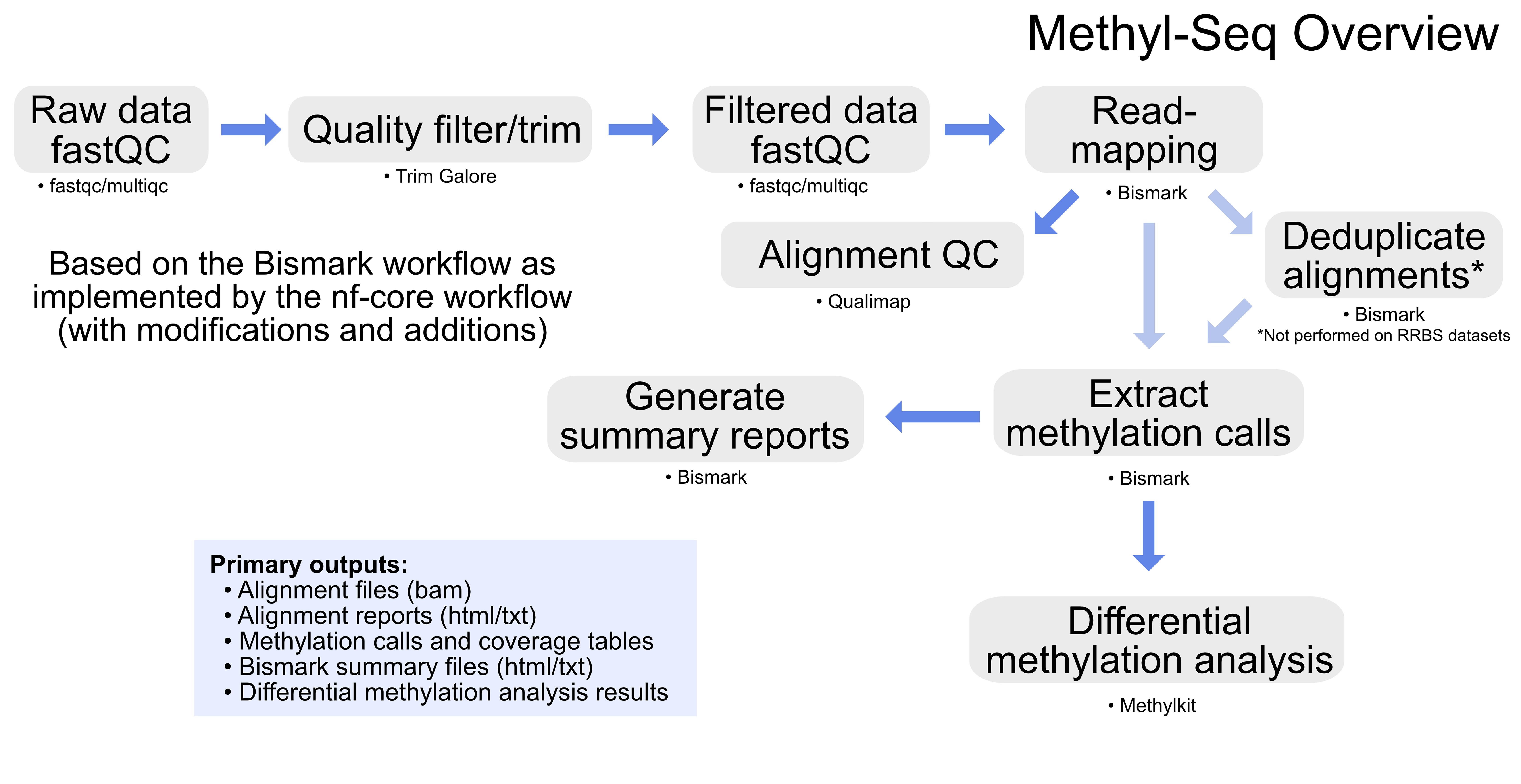
The GeneLab Methyl-Seq consensus processing pipeline is designed to determine how cytosine methylation states change when living organisms are exposed to the space environment by processing raw DNA sequence data, from samples prepared with enzymatic methylation or bisulfite sequencing kits, through differential methylation analysis as summarized in the diagram above.
Specific details of each step of the pipeline, including previous and current pipeline versions, are available on the Methyl-Seq page of the GeneLab DP GitHub Repository. The primary data products and respective quality control (QC) analyses generated from each step of the pipeline, as described below, are available for each GeneLab Methyl-Seq dataset hosted on the OSDR under ‘Study Files’. Below are the Methyl-Seq data files that are published.
Raw sequence data and QC
- *raw.fastq.gz: Raw sequence data, commonly referred to as raw reads
- *raw_multiqc_report.zip: Combined fastQC analyses and respective html report of raw sequence data
Trimmed sequence data and QC
- *trimmed.fastq.gz: Adapter-trimmed and quality-filtered sequence data, commonly referred to as trimmed reads
- *trimming_report.txt: Report detailing the raw read trimming process
- *trimmed_multiqc_report.zip: Combined fastQC analyses and respective html report of trimmed sequence data
Aligned sequence data and QC
- *bismark_bt2_sorted.bam: Sequence alignment map containing reads mapping to the Bismark reference genome (generated using an Ensembl reference genome) and methylation calls, sorted by chromosomal coordinate, in binary format
- *report.txt: Bismark alignment and methylation call report, detailing mapping quality, number of cytosines analyzed, and estimates of methylation calls (CpG, CHG, and CHH)
- *genomic_nucleotide_frequencies.txt: Tab-delimited table containing mono-and di-nucleotide frequencies in the reference genome
- *nucleotide_stats.txt: Tab-delimited table containing sample-specific mono-and di-nucleotide sequence compositions and coverage values compared to genomic compositions
- *bismark_bt2_qualimap.zip: Directory containing several alignment QC data files and a respective HTML report generated using the qualimap bamqc function
Deduplicated aligned sequence data and QC
- *deduplicated.bam: Bismark Bowtie2 alignment bam file sorted by chromosomal coordinate (described above), with duplicates removed
- *deduplication_report.txt: Report file containing deduplication information and statistics
Methylation call data and QC
- *context*txt.gz: gzip-compresed Bismark methylation call files for CpG, CHG, and CHH contexts that were detected, in the Bismark methylation extractor format
- *bedGraph.gz: gzip-compressed file containing methylation percentages of each CpG site, in bedGraph-format
- *bismark.cov.gz: Similar to the gzip-compressed file containing methylation percentages of each CpG site, in bedGraph-format, described above but with 2 additional columns specifying the position of methylated and unmethylated CpGs
- *M-bias.txt: Text file containing methylation information in the context of the position in reads, which can be used to investigate bias as a function of base position in the read as described in the Bismark documentation
- *splitting_report.txt: Text file containing general methylation detection information, including strand-specific methylation information
- *cytosine_context_summary.txt: Text file containing detected cytosine methylation information (from both forward and reverse strands), including their position, strand, trinucleotide content, and methylation state
- *CpG_report.txt.gz: A gzip-compressed, genome-wide methylation report for all CpG cytosines
Alignment and methylation combined reports
- *report.html: Graphical summary of all Bismark alignment, deduplication (for non-RRBS prepared samples), and methylation extraction reports for a single sample, in HTML format
- *bismark_summary_report.txt: Summary table containing information provided in the Bismark alignment, deduplication (for non-RRBS prepared samples), and methylation extraction reports for all samples
- * bismark_summary_report.html: Graphical summary of all information in the *bismark_summary_report.txt file described above for all samples, in HTML format
Annotation files
- *reference.bed: Genome annotation file in BED format
- *reference-gene-to-transcript-map.tsv: Table containing gene-to-transcript mapping with gene IDs in the first column and transcript IDs in the second column
Differential methylation analysis data
Note: Differential methylation analysis is performed using the methylKit R package
- *sig-diff-methylated-bases.tsv: Table containing pairwise analysis of all significantly differentially methylated cytosines
- *sig-diff-hypermethylated-bases.tsv: Table containing pairwise analysis of all cytosines with significantly elevated methylation levels
- *sig-diff-hypomethylated-bases.tsv: Table containing pairwise analysis of all cytosines with significantly reduced methylation levels
- *sig-diff-methylated-tiles.tsv: Table containing pairwise analysis of all significantly differentially methylated tiles
- *sig-diff-hypermethylated-tiles.tsv: Table containing pairwise analysis of all tiles with significantly elevated methylation levels
- *sig-diff-hypomethylated-tiles.tsv: Table containing pairwise analysis of all tiles with significantly reduced methylation levels
- *base-level-percent-methylated.tsv: Table containing the percent methylation levels of all cytosines across all samples
- *tile-level-percent-methylated.tsv: Table containing the percent methylation levels of all tiles across all samples
- *sig-diff-methylated-bases-across-features.pdf: Overview figure containing pairwise analysis of the percent of significantly differentially methylated cytosines identified in specific features (promoter, exon, intron)
- *sig-diff-methylated-tiles-across-features.pdf: Overview figure containing pairwise analysis of the percent of significantly differentially methylated tiles identified in specific features (promoter, exon, intron)
