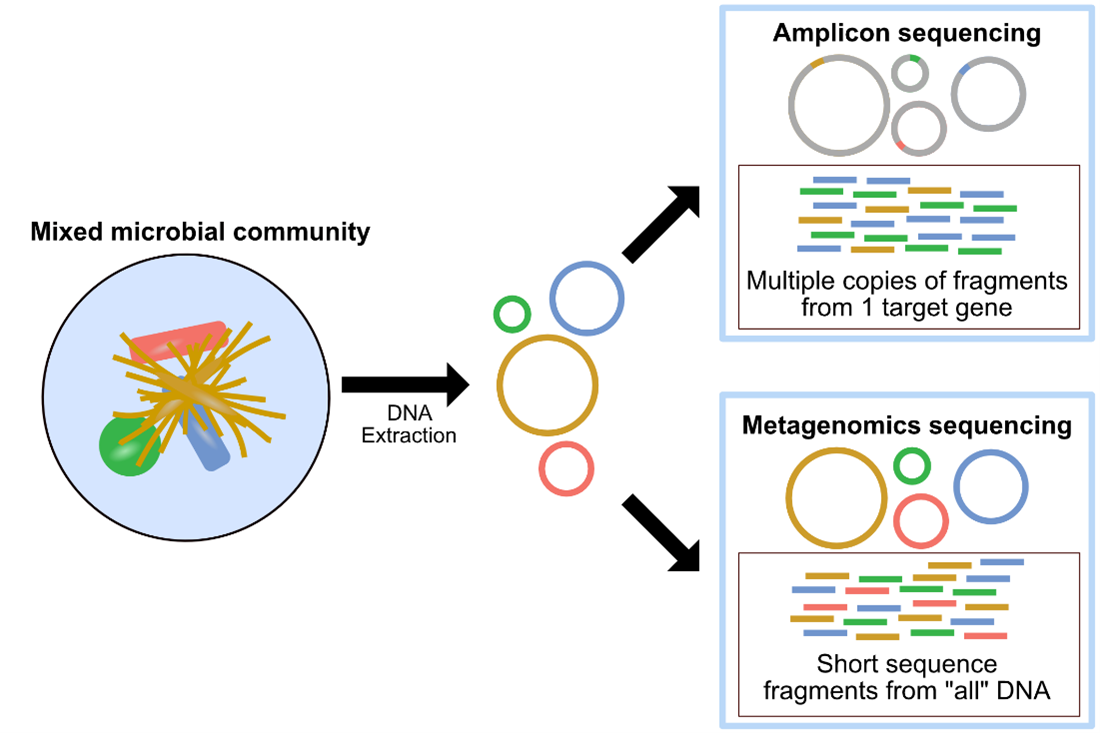
Amplicon sequencing and metagenomics are two commonly used approaches in microbial ecology. The image above, also found here, provides a summary of amplicon sequencing versus metagenomics methodologies. Amplicon sequencing involves using specific primers that target specific genes or gene fragments. These target genes are usually chosen due to their usefulness in delineating their source organisms in a system. Amplicon sequencing is one of the first tools in the microbial ecologist’s toolkit. It is most often used as a broad-level survey of community composition to help generate hypotheses based on differences between recovered sequences or their assigned taxonomies between samples. Traditional methods of processing amplicon data cluster sequences into groups based on percent similarity, most often referred to as Operational Taxonomic Units (OTUs) – while newer approaches take into account things like sequencing error and attempt to infer the original biological sequences, most often referred to as Amplicon Sequence Variants (ASVs). More on the differences between these can be found here.
Some common uses of amplicon sequencing data include:
- providing a metric of community composition based just on the sequences or through assigned taxonomy of those sequences
- tracking changes in community composition in response to a treatment and/or across environmental gradients or time
- providing information on the variety of gene types that exist in a system
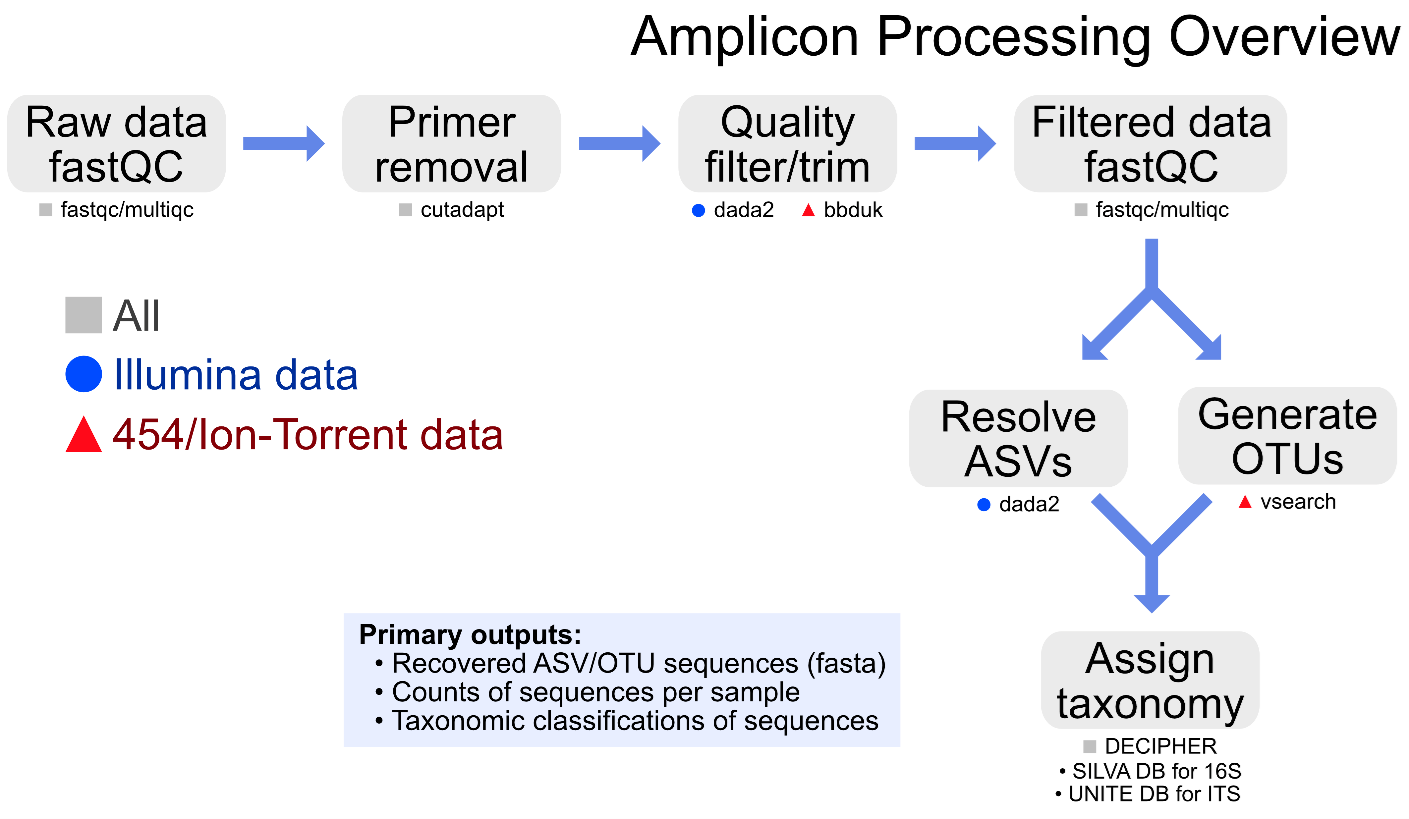
Specific details of each step of the pipeline, including previous and current pipeline versions for both Illumina- and 454/Ion-Torrent-generated data, are available on the Amplicon page of the GeneLab DP GitHub Repository.
The primary data products and respective quality control (QC) analyses generated from each step of the pipeline, as described below, are available for each GeneLab amplicon sequencing dataset hosted on the OSDR under ‘Study Files’. Below are the Amplicon Sequencing published data files:
README
- *readme.txt: A text file describing the output directory structure and data files
Raw sequence data and QC
- *raw.fastq.gz: Raw sequence data, commonly referred to as raw reads
- *raw_multiqc_report.zip: Combined fastQC analyses and respective html report of raw sequence data
Trimmed sequence data and QC
- *trimmed.fastq.gz: Primer-trimmed sequence data, commonly referred to as trimmed reads
- *cutadapt.log: Log file containing the cutadapt trimming report standard output and error
- *trimmed-read-counts.tsv: Per sample read counts before and after trimming
Filtered sequence data and QC
- *filtered.fastq.gz: Quality-filtered sequence data, commonly referred to as filtered reads
- *filtered-read-counts.tsv: Per sample read counts before and after trimming
- *bbduk.log: Log file containing the bbduk filtering report standard output and error (for 454/Ion-Torrent-generated data only)
- *filtered_multiqc_report.zip: Combined fastQC analyses and respective html report of trimmed and filtered sequence data
Final outputs
- *OTUs.fasta: Recovered sequences with singletons and chimeras removed (for 454/Ion-Torrent-generated data only)
- *ASVs.fasta: Recovered sequences (for Illumina-generated data only)
- *read-count-tracking.tsv: Table containing read counts at each processing step
- *counts.tsv: Table containing counts for each recovered sequence
- *taxonomy.tsv: Table containing assigned taxonomy for each recovered sequence
- *taxonomy-and-counts.tsv: Table containing both counts and assigned taxonomy for each recovered sequence
- *taxonomy-and-counts.biom.zip: Zipped file containing both counts and assigned taxonomy for each recovered sequence in biom format
Processing info
- *processing_info.tar: Tarred file containing the exact processing information for each step of the pipeline
