Bulk RNAseq data are derived from Ribonucleic Acid (RNA) molecules that have been isolated from organism cells, tissue(s), organ(s), or a whole organism then reverse transcribed into complimentary Deoxyribonucleic Acid (cDNA) and prepared as libraries to be sequenced on a next generation sequencing platform. There are several different types of RNA molecules including structural RNAs such as ribosomal RNA (rRNA) and transfer RNA (tRNA), non-coding RNAs that regulate gene expression such as long non-coding RNA (lncRNA) and micro RNA (miRNA), and protein-coding RNAs that enable gene expression including messenger RNA (mRNA). Ribosomal RNA is the most abundant type of RNA, making up > 80% of RNA molecules in most cells studied to date. Thus, unless specifically evaluating rRNA, bulk RNAseq studies are designed to investigate changes in gene expression by studying non-rRNA molecules and therefore rRNA is removed during library preparation. The ribo-depletion mechanism to remove rRNA is the standard library preparation method used by GeneLab for in-house bulk RNAseq data generation because this approach removes rRNA, allowing all other types of RNA to be sequenced and subsequently analyzed. Another commonly used approach is to positively select for mRNA by targeting the polyA tail at the 3’ end of mRNA molecules, a mechanism referred to as polyA-selection, which only allows for sequencing and evaluation of mRNA.
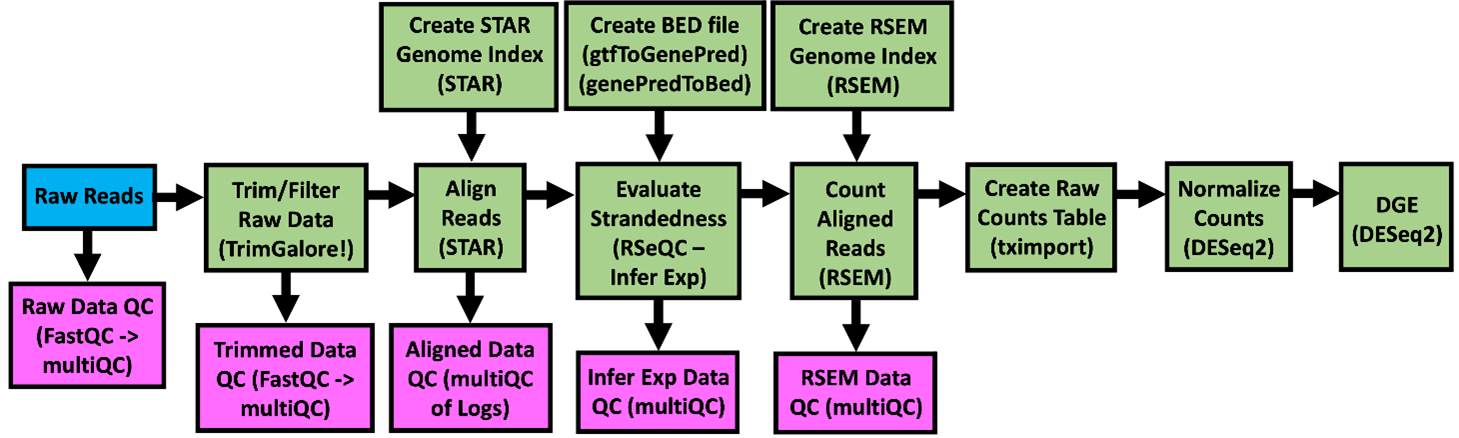
The GeneLab bulk RNAseq consensus processing pipeline (RCP), is designed to determine how gene expression changes (i.e. which genes are turned on, turned off, or stay the same) when living organisms are exposed to the space environment by processing raw RNA sequence data through differential gene expression as summarized in the diagram above. Specific details of each step of the pipeline, including previous and current pipeline versions, are available on the RNAseq page of the GeneLab DP GitHub Repository.
The primary data products and respective quality control (QC) analyses generated from each step of the pipeline, as described below, are available for each GeneLab RNAseq dataset hosted on the OSDR under ‘Study Files’. Below are the bulk RNAseq data files that are published:
Raw sequence data and QC
- *raw.fastq.gz: Raw sequence data, commonly referred to as raw reads
- *raw_multiqc_report.zip: Combined fastQC analyses and respective html report of raw sequence data
Trimmed sequence data and QC
- *trimmed.fastq.gz: Trimmed and quality-filtered sequence data, commonly referred to as trimmed reads
- *trimming_report.txt: Report detailing the raw read trimming process
- *trimmed_multiqc_report.zip: Combined fastQC analyses and respective html report of trimmed sequence data
Aligned sequence data and QC
- *Aligned.sortedByCoord_sorted.out.bam: Sequence alignment map containing reads mapping to the Ensembl reference genome, sorted by coordinate, in binary format
- *Aligned.toTranscriptome.out.bam: Sequence alignment map containing reads mapping to the Ensembl reference transcripts, in binary format
- *SJ.out.tab: Tab-deliminted file containing identified splice junctions and respective count data
- *Log.final.out: Log file containing alignment info/stats such as the number of uniquely mapped reads and multi-mapped reads
- *align_multiqc_report.zip: Combined alignment info/stats data from each sample and respective html report
RSeQC reports
- *infer_exp_multiqc_report.zip: Combined RSeQC infer_experiment data, specifying read strandedness, from each sample and respective html report
- *geneBody_cov_multiqc_report.zip: Combined RSeQC geneBody_coverage data, quantifying relative 5’ to 3’ transcript coverage, from each sample and respective html report
- *read_dist_multiqc_report.zip: Combined RSeQC read_distribution data, identifying relative abundance of each feature type, from each sample and respective html report
- *inner_dist_multiqc_report.zip: Combined RSeQC inner_distance data, indicating the distance (in number of bases) between the forward and respective reverse reads, from each sample and respective html report (for paired-end data only). Note: Negative values indicate overlap of forward and respective reverse reads
Raw counts data and QC
- *genes.results: Expression estimates per gene in RSEM expected raw counts, transcripts per million (TPM), and fragments per kilobase of transcript per million mapped reads (FPKM)
- *isoforms.results: Expression estimates per isoform in RSEM expected raw counts, transcripts per million (TPM), and fragments per kilobase of transcript per million mapped reads (FPKM)
- *RSEM_count_multiqc_report.zip: Combined alignment info/stats data used for RSEM quantification from each sample and respective html report
- *RSEM_Unnormalized_Counts.csv: Table containing expected raw counts for each sample quantified with RSEM
- *STAR_Unnormalized_Counts.csv: Table containing raw counts for each sample quantified with STAR
Normalized counts data
- *Normalized_Counts.csv: Table containing normalized counts using the DESeq2 median of ratios method
- *ERCC_Normalized_Counts.csv (available upon request): Table containing normalized counts using the DESeq2 median of ratios method in which only group B ERCC genes are used for size factor estimation (only included for datasets with samples containing ERCC spike-in)
Differential expression analysis data
- *SampleTable.csv: Table containing sample names and their respective groups
- *contrasts.csv: Table containing all pair-wise group comparisons
- *differential_expression.csv: Table containing normalized counts for each sample, group statistics, DESeq2 differential gene expression (DGE) results for each pairwise comparison, and gene annotations
- *ERCCnorm_SampleTable.csv (available upon request): Table containing the names of each sample with detectable ERCC group B genes and their respective groups (only included for datasets with samples containing ERCC spike-in)
- *ERCCnorm_contrasts.csv (available upon request): Table containing pair-wise group comparisons for groups containing samples with detectable ERCC group B genes (only included for datasets with samples containing ERCC spike-in)
- *ERCCnorm_differential_expression.csv (available upon request): Table containing ERCC-normalized counts for each sample, group statistics, DESeq2 differential gene expression (DGE) results for each pairwise comparison (using ERCC-normalized counts), and gene annotations (only included for datasets with samples containing ERCC spike-in)
ERCC analyses (only included for datasets with samples containing ERCC spike-in)
- *ERCC_analysis*.html: Completed ERCC spike-in analysis Jupyter Notebook in html format
